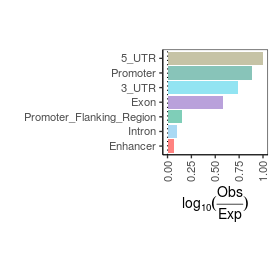
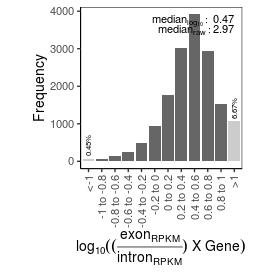
Sample name: K562_RNA-seq_30
Looper stats summary
| sample_name |
K562_RNA-seq_30 |
| sample_desc |
70% K562 PRO-seq + 30% K562 RNA-seq |
| treatment |
70M total reads |
| protocol |
PRO |
| organism |
human |
| read_type |
SINGLE |
| umi_len |
0 |
| read1 |
/project/shefflab/data//guertin/fastq/K562_30pctRNA.fastq.gz |
| srr |
K562_30pctRNA |
| Raw_reads |
70000000 |
| Fastq_reads |
70000000 |
| Reads_with_adapter |
49200456.0 |
| Uninformative_adapter_reads |
1225550.0 |
| Pct_uninformative_adapter_reads |
1.7508 |
| Trimmed_reads |
68774450 |
| Trim_loss_rate |
1.75 |
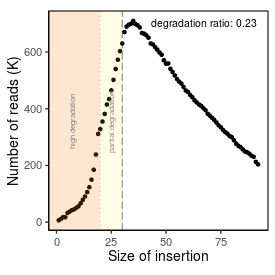
| Peak_adapter_insertion_size |
34 |
| Degradation_ratio |
0.231 |
| Aligned_reads_human_rDNA |
4894873.0 |
| Alignment_rate_human_rDNA |
7.12 |
| Mapped_reads |
59888336 |
| QC_filtered_reads |
8412839 |
| Aligned_reads |
51475497 |
| Alignment_rate |
74.85 |
| Total_efficiency |
73.54 |
| Read_depth |
6.15 |
| Mitochondrial_reads |
2160789 |
| Maximum_read_length |
100 |
| Genome_size |
3099922541 |
| NRF |
0.77 |
| PBC1 |
0.9 |
| PBC2 |
13.32 |
| Unmapped_reads |
3991241 |
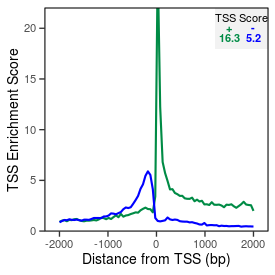
| TSS_coding_score |
16.3 |
| TSS_non-coding_score |
4.9 |
| File_mb |
5620.96 |
| Read_type |
SINGLE |
| Genome |
hg38 |
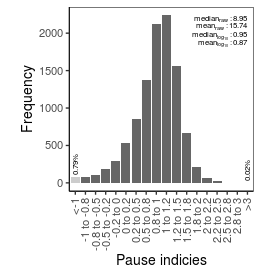
| Pause_index |
8.95 |
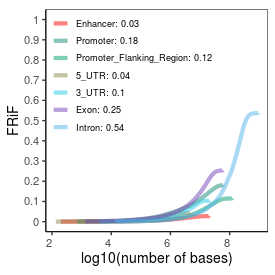
| Plus_FRiP |
0.37 |
| Minus_FRiP |
0.36 |
| mRNA_contamination |
2.97 |
| Time |
0:36:29 |
| Success |
06-14-21:47:46 |